
El estudio ha sido recibido: 26 de abril de 2018 y aceptado: 16 de julio de 2018.
Publicado en línea: 09 de agosto de 2018
Nilambra Dogra1,2, Ashok Kumar1,3 y Tapas Mukhopadhyay1
1 – Centro Nacional de Estudios e Investigación del Genoma Humano, Universidad Panjab, Sector-14, Chandigarh, 160014, India. 2 – Dirección actual: Department of Experimental Medicine and Biotechnology, Postgraduate Institute of Medical Education and Research, Sector-12, Chandigarh, 160012, India. 3 – Dirección actual: Centro de Biología de Sistemas y Bioinformática, Universidad Panjab, Sector-25, Chandigarh, 160014, India. La correspondencia y las solicitudes de material deben dirigirse a T.M. (correo electrónico: [email protected])
Los fármacos que ya han sido aprobados clínicamente o probados experimentalmente para enfermedades distintas del cáncer, pero que poseen una citotoxicidad previamente desconocida contra las células malignas, pueden servir como candidatos adecuados contra el cáncer. El N-(6-fenilsulfanil-1H benzimidazol-2-il) carbamato de metilo [Fenbendazol, FZ], un compuesto de benzimidazol, es un fármaco antihelmíntico seguro y barato que posee una eficaz actividad antiproliferativa.
En nuestro trabajo anterior, informamos de una potente actividad inhibidora del crecimiento de FZ causada en parte por el deterioro de la función proteasomal. Aquí demostramos que el FZ muestra una afinidad moderada por la tubulina de mamífero y ejerce citotoxicidad en células cancerosas humanas a concentraciones micromolares. Simultáneamente, provocó la translocación mitocondrial de p53 e inhibió eficazmente la captación de glucosa, la expresión de los transportadores GLUT, así como la hexoquinasa (HK II), una enzima glucolítica clave de la que se nutren la mayoría de las células cancerosas. Bloqueó el crecimiento de xenoinjertos humanos en el modelo de ratón nu/nu cuando los ratones fueron alimentados con el fármaco por vía oral.
Los resultados, junto con nuestros datos anteriores, sugieren que FZ es un nuevo agente interferente de los microtúbulos que muestra actividad antineoplásica y puede evaluarse como posible agente terapéutico por su efecto en múltiples vías celulares que conduce a la eliminación eficaz de las células cancerosas.
La importancia de los microtúbulos en la división celular, la motilidad, el tráfico intracelular y su papel en la modulación de la forma celular en función del entorno los ha convertido en una de las dianas más exitosas de la terapia anticancerosa. Los agentes que perturban la dinámica de los microtúbulos se han utilizado ampliamente en el tratamiento del cáncer1-4. Teniendo en cuenta el relativo éxito de los agentes mitóticos en el tratamiento del cáncer, los microtúbulos pueden considerarse una de las mejores dianas contra el cáncer identificadas hasta la fecha5.
Los agentes dirigidos a los microtúbulos pueden clasificarse en dos grandes categorías. La primera clase consiste en agentes desestabilizadores de microtúbulos, que inhiben la polimerización de los microtúbulos. Esta clase de fármacos antimitóticos incluye varios compuestos como los alcaloides de la vinca (vinblastina, vincristina, vinorelbina, vindesina, vinflunina), la estramustina, la colchicina y las combretastatinas, que se están utilizando clínicamente o se están investigando clínicamente para el tratamiento del cáncer. La segunda clase se compone de agentes estabilizadores de microtúbulos. Estos agentes incluyen paclitaxel, docetaxel, epothilones y discodermolide6.
La consecuencia de alterar la dinámica de la tubulina y los microtúbulos con estas dos clases de fármacos en las células en división es la detención de la metafase y la inducción de la apoptosis.
El fenbendazol (metil N-(6-fenilsulfanil-1H-benzimidazol-2-il) carbamato) es un antihelmíntico benzimidazólico de amplio espectro aprobado para su uso en numerosas especies animales7. La reutilización para uso humano de medicamentos veterinarios con resultados prometedores puede reducir considerablemente el tiempo y los costes necesarios para desarrollar nuevos fármacos. Se sabe que el fenbendazol tiene un alto margen de seguridad y que la mayoría de las especies lo toleran muy bien. Tiene muy bajo grado de toxicidad y alto grado de seguridad en animales de experimentación8-12.
En este estudio, demostramos que el fenbendazol FZ) presenta una moderada actividad despolimerizadora de los microtúbulos frente a las células cancerosas humanas, pero posee un potente efecto antitumoral, como se desprende de los experimentos in vitro e in vivo. Nuestros resultados indican que FZ ejerce su efecto antitumoral a través de la interrupción de la dinámica de los microtúbulos, la activación de p53 y la modulación de genes implicados en múltiples vías celulares. El tratamiento con FZ también redujo la captación de glucosa en las células cancerosas debido a la regulación a la baja de los transportadores GLUT y las enzimas glucolíticas clave.
Dado que en el proceso de tumorigénesis intervienen varios genes y proteínas que alteran diversas vías de señalización celular, los fármacos dirigidos a una única diana muestran una eficacia limitada y pueden provocar resistencia a los medicamentos13-15. Por lo tanto, se espera que los agentes con múltiples dianas celulares tengan una eficacia mejorada, además de la capacidad de eludir la probabilidad de desarrollar resistencia. En general, el presente trabajo demuestra un efecto pleiotrópico de FZ sobre las células cancerosas que conduce a la muerte celular. Así pues, FZ puede tener una aplicación terapéutica potencial.
Resultados
La FZ desestabiliza la red de tubulina en células humanas de CPNM.
Se ha descrito que los carbamatos de benzimidazol inhiben la polimerización de la tubulina y alteran la función de los microtúbulos en células parasitarias16,17. Los resultados de estudios in vitro con extractos enriquecidos de tubulina de helmintos y mamíferos han sugerido que la tubulina es la diana molecular principal de los benzimidazoles18.
Por lo tanto, para examinar el efecto de FZ en la organización de la red de microtúbulos de mamíferos, se trataron células A549 de carcinoma pulmonar humano no microcítico (CPNM) con 1 uM de FZ durante 24 h y se procesaron para inmunofluorescencia utilizando el anticuerpo α-tubulina. La colchicina se utilizó como control positivo. Los resultados mostraron que el tratamiento con FZ causó una alteración parcial de la red de microtúbulos (Fig. 1a). La jaula de microtúbulos alrededor del núcleo parecía haber perdido su integridad en comparación con las células de control tratadas con simulacro. Sin embargo, esta modificación en la organización no fue tan marcada como en el caso del tratamiento con colchicina, que mostró una despolimerización completa de los microtúbulos en subunidades de tubulina. Estos datos sugieren que la FZ provoca una distorsión del entramado microtubular de las células.
El efecto de la FZ sobre la polimerización de la tubulina se evaluó además mediante un ensayo in vitro. Se incubó tubulina bovina purificada con FZ y se registró la polimerización de la tubulina a lo largo del tiempo. Los resultados mostraron una leve inhibición de la polimerización de tubulina por FZ in vitro, que no fue tan pronunciada como en el caso del tratamiento con colchicina. (Fig. 1b)
A continuación, se comparó el efecto de FZ sobre la polimerización de tubulina con el de otros agentes desestabilizadores de microtúbulos como el nocodazol y la colchicina. Se prepararon fracciones polimerizadas y solubles tras 24 h de tratamiento farmacológico y se realizó western blot utilizando anticuerpos α-tubulina y β-actina (Fig. 1c). Las bandas de tubulina de las fracciones polimerizadas y solubles se cuantificaron tras normalizarlas con sus respectivas bandas de β-actina, que sirvieron como control interno (Fig. 1d).
Hubo una modesta disminución de la tubulina polimérica en las células tratadas con FZ en comparación con las células control no tratadas, mientras que la forma polimerizada de la tubulina estaba casi ausente en las células tratadas con colchicina. El resultado confirma la actividad despolimerizadora de la tubulina relativamente suave de FZ en comparación con otros agentes disruptores de microtúbulos conocidos como el nocodazol y la colchicina.
Uno de los principales factores limitantes de los taxanos y los alcaloides de la vinca es su toxicidad dosis-limitante y su susceptibilidad a la resistencia a múltiples fármacos (MDR), que suele deberse a la elevada expresión de la p-glicoproteína (p-gp; MDR1)19,20 . También se sabe que la sobreexpresión de isoformas de β-tubulina y las mutaciones confieren resistencia a los taxanos21.
A diferencia de los taxanos y los alcaloides de la vinca, los fármacos dirigidos al sitio de unión de la colchicina presentan la ventaja de mostrar una resistencia mínima a múltiples fármacos, además de su capacidad para superar el efecto de la sobreexpresión de isoformas de β-tubulina22-24. Sin embargo, el principal inconveniente de los fármacos dirigidos a la colchicina es su capacidad para superar el efecto de la sobreexpresión de isoformas de β-tubulina. Sin embargo, el principal inconveniente de la colchicina y sus derivados es su toxicidad aguda para el ser humano22,25. Por lo tanto, un inhibidor de los microtúbulos que se una al sitio de unión de la colchicina pero que tenga una toxicidad baja puede ser muy eficaz26,27. El resultado de un ensayo de unión competitiva a colchicina basado en fluorescencia sugiere que FZ puede unirse a la tubulina en el sitio de unión a colchicina (Fig. S1).
La acetilación de la tubulina se ha asociado con la estabilidad de los microtúbulos. Por lo tanto, para examinar el estado de acetilación de la tubulina tras el tratamiento, las células humanas NSCLC fueron tratadas con diferentes agentes dirigidos a los microtúbulos durante 24 h y los extractos celulares fueron sometidos a análisis western blot utilizando el anticuerpo específico Ac-α-tubulina (6-11B-1). Como se muestra en la Fig. 1e, mientras que el nocodazol, la colchicina y la vincristina produjeron una marcada reducción de la tubulina acetilada, FZ no alteró la cantidad de tubulina acetilada en comparación con las células tratadas con simulacros de control. Este resultado confirmó además el efecto relativamente leve de FZ sobre la tubulina de mamíferos en comparación con otros agentes despolimerizadores de microtúbulos conocidos.
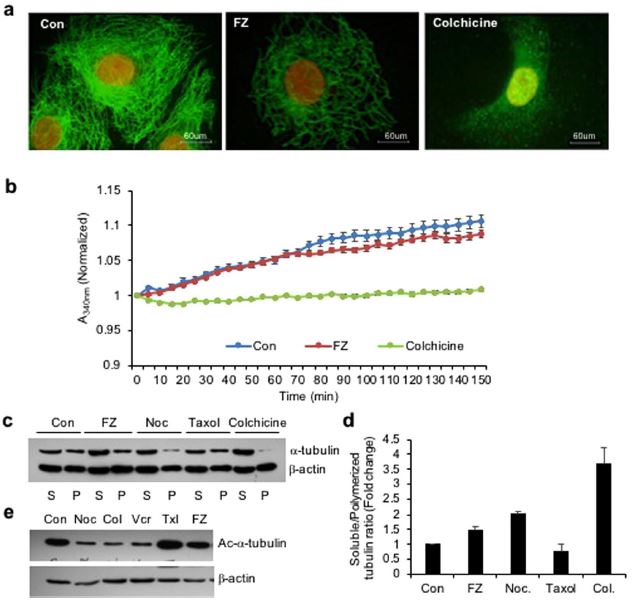
Figura 1. El tratamiento con FZ altera la red de tubulina de las células cancerosas humanas. (a) Las células A549 se trataron con 1 uM de FZ o 50 ng/ml de colchicina durante 24 h. Tras el tratamiento, las células se procesaron para inmunofluorescencia utilizando anticuerpos primarios anti α-tubulina y secundarios conjugados con FITC. (Los núcleos se contrateñían con yoduro de propidio) (b) la tubulina bovina (1,8 mg/mL) se incubó con DMSO (control), FZ (10 uM) o colchicina (100 nM) y el efecto sobre la polimerización se controló espectrofotométricamente midiendo la turbidez a 340 nm como se describe en «Métodos.» (c) Las células se trataron con FZ, nocodazol, taxol o colchicina durante 24 h y después se lisaron y fraccionaron en extractos solubles (S) y polimerizados (P). Los extractos se separaron mediante SDS-PAGE, se transfirieron a membranas de PVDF y se analizaron con anticuerpos anti-α-tubulina y anti-β-actina. Se muestra un análisis de inmunotransferencia representativo en células A549. (d) La intensidad de cada banda del inmunoblot se midió con el programa NIH ImageJ, y se calcularon las proporciones de tubulina soluble y polimerizada y de β-actina en cada tratamiento. (e) Las células se trataron con diferentes MTA según se indica durante 24 h y a continuación se realizó el western blot utilizando anticuerpos específicos de Ac-α-tubulina (6-11B-1) y β-actina. (Los blots completos sin recortar se incluyen en la Fig. suplementaria S6).
FZ no es un sustrato o inhibidor de la P-gp.
El desarrollo de farmacorresistencia es una de las principales preocupaciones en el tratamiento del cáncer. La resistencia a múltiples fármacos (MDR) causada por la sobreexpresión del gen MDR-1, que codifica la glicoproteína P (P-gp), es un mecanismo crítico de resistencia a fármacos que da lugar a una resistencia cruzada a múltiples clases de fármacos28,29 .
Un gran número de fármacos quimioterápicos de uso común, como los taxanos y los alcaloides de la vinca, son sustratos de la P-gp30.
Sin embargo, los esfuerzos por inhibir la P-gp no han dado resultados alentadores debido a los inevitables efectos secundarios31,32. Por lo tanto, el descubrimiento y desarrollo de nuevos compuestos antiproliferativos que no sean sustratos de la P-gp es un enfoque eficaz para superar la resistencia a los fármacos. Para comprobar si FZ es un sustrato o un inhibidor de la P-gp, investigamos la inhibición del crecimiento de células cancerosas por FZ en presencia del inhibidor de la P-gp verapamilo. Los resultados mostraron que la inhibición de P-gp por verapamilo no potenció el efecto inhibidor de FZ sobre la proliferación de células cancerosas (Fig. 2c). El colorante fluorescente rodamina 123 (Rho123) es un sustrato de P-gp de referencia bien conocido que se utiliza con frecuencia para determinar el potencial inhibidor de P-gp de los fármacos33.
No se observó ninguna diferencia significativa en la acumulación de Rho123 entre las células de control no tratadas y las tratadas con FZ, lo que implica la ausencia de cualquier interacción de FZ con la P-gp. (Fig. 2a,b) En presencia de verapamilo, las células tratadas y no tratadas mostraron niveles comparables de acumulación de Rho123 afirmando que FZ no es un sustrato o inhibidor de P-gp.
{kind=link}