
The study has been received: 26 April 2018 and accepted: 16 July 2018.
Published online: 09 August 2018
Nilambra Dogra1,2, Ashok Kumar1,3 & Tapas Mukhopadhyay1
1 – National Centre for Human Genome Studies and Research, Panjab University, Sector-14, Chandigarh, 160014, India. 2 – Present address: Department of Experimental Medicine and Biotechnology, Postgraduate Institute of Medical Education and Research, Sector-12, Chandigarh, 160012, India. 3 – Present address: Centre for Systems Biology and Bioinformatics, Panjab University, Sector-25, Chandigarh, 160014, India. Correspondence and requests for materials should be addressed to T.M. (email: [email protected])
Drugs that are already clinically approved or experimentally tested for conditions other than cancer, but are found to possess previously unrecognized cytotoxicity towards malignant cells, may serve as fitting anti-cancer candidates. Methyl N-(6-phenylsulfanyl-1H benzimidazol-2-yl) carbamate [Fenbendazole, FZ], a benzimidazole compound, is a safe and inexpensive anthelmintic drug possessing an efficient anti-proliferative activity.
In our earlier work, we reported a potent growth-inhibitory activity of FZ caused partially by impairment of proteasomal function. Here, we show that FZ demonstrates moderate affinity for mammalian tubulin and exerts cytotoxicity to human cancer cells at micromolar concentrations. Simultaneously, it caused mitochondrial translocation of p53 and effectively inhibited glucose uptake, expression of GLUT transporters as well as hexokinase (HK II) – a key glycolytic enzyme that most cancer cells thrive on. It blocked the growth of human xenografts in nu/nu mice model when mice were fed with the drug orally.
The results, in conjunction with our earlier data, suggest that FZ is a new microtubule interfering agent that displays anti-neoplastic activity and may be evaluated as a potential therapeutic agent because of its effect on multiple cellular pathways leading to effective elimination of cancer cells.
The importance of microtubules in cell division, motility, intracellular trafficking and their role in modulating cellular shape according to the environment has made them one of the most successful targets of anticancer therapy. Agents that perturb the microtubule dynamics have been widely used in cancer treatment1-4. Considering the relative success of mitotic agents in the treatment of cancer, microtubules may be termed as one of the best cancer targets identified till now5.
Microtubule targeting agents can be broadly classified into two major classes. The first class consists of microtubule-destabilizing agents, which inhibit microtubule polymerization. This class of anti-mitotic drugs includes several compounds such as the vinca alkaloids (vinblastine, vincristine, vinorelbine, vindesine, vinflunine), estramustine, colchicine and combretastatins, that are being used clinically or are under clinical investigation for cancer treatment. The second class is comprised of microtubule-stabilizing agents. These agents include paclitaxel, docetaxel, epothilones, and discodermolide6.
The consequence of disrupting tubulin and microtubule dynamics with both these classes of drugs in dividing cells is metaphase arrest and induction of apoptosis.
Fenbendazole (methyl N-(6-phenylsulfanyl-1H-benzimidazol-2-yl) carbamate) is a broad-spectrum benzimidazole anthelminthic approved for use in numerous animal species7. Repurposing of veterinary drugs showing promising results for human use can result in considerable time and cost reduction required to develop new drugs. Fenbendazole is known to have a high safety margin and most species tolerate it very well. It has very low degree of toxicity and high degree of safety in experimental animals8–12.
In this study, we show that fenbendazole FZ) exhibits a moderate microtubule depolymerizing activity towards human cancer cells, but possesses a potent antitumor effect as evident from in vitro and in vivo experiments. Our results indicate that FZ exerts its antitumor effect through the disruption of microtubule dynamics, p53 activation and the modulation of genes involved in multiple cellular pathways. FZ treatment also resulted in reduced glucose uptake in cancer cells due to down regulation of GLUT transporters and key glycolytic enzymes.
Since the process of tumorigenesis involves a number of genes and proteins altering various cell signaling pathways, single-target drugs show limited efficacy and may lead to drug resistance13–15. Agents having multiple cellular targets, therefore, are expected to have improved efficacy besides the ability to circumvent the likelihood of developing resistance. Overall, the present work demonstrates a pleiotropic effect of FZ on cancer cells leading to cell death. Thus, FZ may have a potential therapeutic application.
Results
FZ destabilizes tubulin network in human NSCLC cells. Benzimidazole carbamates have been reported to inhibit tubulin polymerization and disrupt microtubule function in parasite cells16,17.Results from in vitro studies using enriched extracts of helminthic and mammalian tubulin have suggested that tubulin is the primary molecular target of the benzimidazoles18.
Therefore, to examine the effect of FZ on mammalian microtubule network organization, human non small cell lung carcinoma (NSCLC) A549 cells were treated with 1 uM FZ for 24 h and processed for immunofluorescence using α-tubulin antibody. Colchicine was used as a positive control. Results showed that FZ treatment caused a partial alteration of the microtubule network (Fig. 1a). The microtubule cage around the nucleus appeared to have lost its intactness when compared with the control mock treated cells. However, this modification in the organization was not as marked as in case of colchicine treatment, which showed complete depolymerization of microtubules into tubulin subunits. This data suggests that FZ causes distorted microtubule framework of the cells.
The effect of FZ on tubulin polymerization was further evaluated by an in vitro assay. Purified bovine tubulin was incubated with FZ, and tubulin polymerization was recorded over time. The results showed mild inhibition of tubulin polymerization by FZ in vitro which was not as pronounced as in case of colchicine treatment. (Fig. 1b)
Next, the effect of FZ on tubulin polymerization was compared with that of other microtubule destabilizing agents like nocodazole and colchicine. Polymerized and soluble fractions were prepared after 24 h drug treatment and western blot was performed using α-tubulin and β-actin antibodies (Fig. 1c). Tubulin bands of polymerized and soluble fractions were quantified after normalization with their respective β-actin bands which served as an internal control (Fig. 1d).
There was a modest decrease in polymeric tubulin in FZ treated cells as compared to control untreated cells, whereas polymerized form of tubulin was nearly absent in colchicine treated cells. The result confirms the relatively mild tubulin depolymerising activity of FZ as compared to other known microtubule disrupting agents like nocodazole and colchicine.
A major limiting factor of taxanes and vinca alkaloids is their dose-limiting toxicity and susceptibility to multidrug drug-resistance (MDR) occurring commonly due to the high expression of p-glycoprotein (p-gp; MDR1)19,20. Overexpression of β-tubulin isoforms and mutations are also known to confer resistance to taxanes21.
Unlike taxanes and vinca alkaloids, agents targeting the colchicine-binding site are advantageous in that they show minimal multidrug resistance in addition to their ability to overcome the effect of β-tubulin isoforms overexpression22–24. However, the major drawback of colchicine and its derivatives is their acute toxicity to humans22,25. Therefore, a microtubule inhibitor that binds to the colchicine-binding site but has low toxicity can be highly efficacious26,27. The result of a fluorescence based competitive colchicine binding assay suggests that FZ may bind to the tubulin at the colchicine binding site (Fig. S1).
Tubulin acetylation has been associated with the stability of microtubules. Therefore, to examine the acetylation status of tubulin following treatment, human NSCLC cells were treated with different microtubule targeting agents for 24 h and the cell extracts were subjected to western blot analysis using Ac-α-tubulin specific antibody (6–11B-1). As shown in Fig. 1e, while nocodazole, colchicine and vincristine resulted in a marked reduction of acetylated tubulin, FZ did not alter the amount of acetylated tubulin as compared with control mock treated cells. This result further confirmed the relatively mild effect of FZ on mammalian tubulin as compared with other known microtubule depolymerizing agents.
 Figure 1. FZ treatment alters tubulin network of human cancer cells. (a) A549 cells were treated with 1 uM FZ or 50 ng/ml colchicine for 24 h. Following treatment, the cells were processed for immunofluorescence using anti α-tubulin primary and FITC conjugated secondary antibodies. (Nuclei were counter stained with propidium iodide) (b) bovine tubulin (1.8 mg/mL) was incubated with DMSO (control), FZ (10 uM) or colchicine (100 nM) and the effect on polymerization was monitored spectrophotometrically by measuring turbidity at 340 nm as described under “Methods.” (c) Cells were treated with FZ, nocodazole, taxol or colchicine for 24 h and then lysed and fractionated into soluble (S) and polymerized (P) extracts. The extracts were separated with SDS-PAGE, transferred onto PVDF membranes and probed with both anti-α-tubulin and anti-β-actin antibodies. A representative immunoblot analysis in A549 cells is shown. (d) Intensity of each band of the immunoblot was measured by the NIH ImageJ program, and the ratios of soluble and polymerized tubulin and β-actin in each treatment were calculated. (e) Cells were treated with different MTAs as indicated for 24 h and western blotting was then performed using Ac-α-tubulin (6–11B-1) specific and β-actin antibodies. (Full-length uncropped blots are included in Supplementary Fig. S6).
FZ is not a P-gp substrate or inhibitor. Development of drug resistance is a major concern in cancer treatment. Multidrug resistance (MDR) caused by the overexpression of the MDR-1 gene that encodes P-glycoprotein (P-gp) is a critical mechanism of drug resistance which results in a cross-resistance to multiple classes of drugs28,29.
A large number of commonly used chemotherapy drugs like taxanes and vinca alkaloids are P-gp substrates30.
However, efforts to inhibit P-gp have not shown encouraging results due to unavoidable side effects31,32. Therefore, discovery and development of novel anti-proliferative compounds that are not substrates of P-gp is an effective approach to overcome drug resistance. To test whether FZ is a substrate or inhibitor of P-gp, we investigated cancer cell growth inhibition by FZ in the presence of P-gp inhibitor verapamil. The results showed that inhibition of P-gp by verapamil did not enhance the inhibitory effect of FZ on cancer cell proliferation (Fig. 2c). The fluorescent dye rhodamine 123 (Rho123) is a well-known reference P-gp substrate frequently used to determine the P-gp inhibitory potential of drugs33.
No significant difference in Rho123 accumulation was observed between control untreated and FZ treated cells, implying the absence of any interaction of FZ with P-gp. (Fig. 2a,b) In the presence of verapamil, the treated and untreated cells showed comparable levels of Rho123 accumulation affirming that FZ is not a substrate or inhibitor of P-gp.
Figure 1. FZ treatment alters tubulin network of human cancer cells. (a) A549 cells were treated with 1 uM FZ or 50 ng/ml colchicine for 24 h. Following treatment, the cells were processed for immunofluorescence using anti α-tubulin primary and FITC conjugated secondary antibodies. (Nuclei were counter stained with propidium iodide) (b) bovine tubulin (1.8 mg/mL) was incubated with DMSO (control), FZ (10 uM) or colchicine (100 nM) and the effect on polymerization was monitored spectrophotometrically by measuring turbidity at 340 nm as described under “Methods.” (c) Cells were treated with FZ, nocodazole, taxol or colchicine for 24 h and then lysed and fractionated into soluble (S) and polymerized (P) extracts. The extracts were separated with SDS-PAGE, transferred onto PVDF membranes and probed with both anti-α-tubulin and anti-β-actin antibodies. A representative immunoblot analysis in A549 cells is shown. (d) Intensity of each band of the immunoblot was measured by the NIH ImageJ program, and the ratios of soluble and polymerized tubulin and β-actin in each treatment were calculated. (e) Cells were treated with different MTAs as indicated for 24 h and western blotting was then performed using Ac-α-tubulin (6–11B-1) specific and β-actin antibodies. (Full-length uncropped blots are included in Supplementary Fig. S6).
FZ is not a P-gp substrate or inhibitor. Development of drug resistance is a major concern in cancer treatment. Multidrug resistance (MDR) caused by the overexpression of the MDR-1 gene that encodes P-glycoprotein (P-gp) is a critical mechanism of drug resistance which results in a cross-resistance to multiple classes of drugs28,29.
A large number of commonly used chemotherapy drugs like taxanes and vinca alkaloids are P-gp substrates30.
However, efforts to inhibit P-gp have not shown encouraging results due to unavoidable side effects31,32. Therefore, discovery and development of novel anti-proliferative compounds that are not substrates of P-gp is an effective approach to overcome drug resistance. To test whether FZ is a substrate or inhibitor of P-gp, we investigated cancer cell growth inhibition by FZ in the presence of P-gp inhibitor verapamil. The results showed that inhibition of P-gp by verapamil did not enhance the inhibitory effect of FZ on cancer cell proliferation (Fig. 2c). The fluorescent dye rhodamine 123 (Rho123) is a well-known reference P-gp substrate frequently used to determine the P-gp inhibitory potential of drugs33.
No significant difference in Rho123 accumulation was observed between control untreated and FZ treated cells, implying the absence of any interaction of FZ with P-gp. (Fig. 2a,b) In the presence of verapamil, the treated and untreated cells showed comparable levels of Rho123 accumulation affirming that FZ is not a substrate or inhibitor of P-gp.
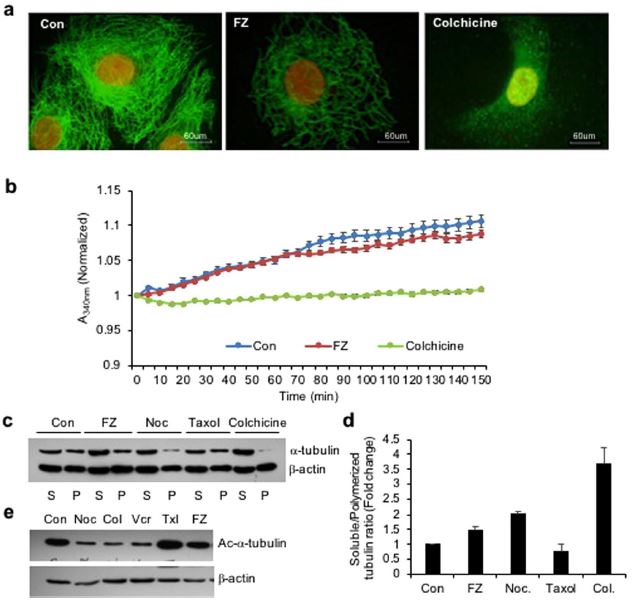 Figure 1. FZ treatment alters tubulin network of human cancer cells. (a) A549 cells were treated with 1 uM FZ or 50 ng/ml colchicine for 24 h. Following treatment, the cells were processed for immunofluorescence using anti α-tubulin primary and FITC conjugated secondary antibodies. (Nuclei were counter stained with propidium iodide) (b) bovine tubulin (1.8 mg/mL) was incubated with DMSO (control), FZ (10 uM) or colchicine (100 nM) and the effect on polymerization was monitored spectrophotometrically by measuring turbidity at 340 nm as described under “Methods.” (c) Cells were treated with FZ, nocodazole, taxol or colchicine for 24 h and then lysed and fractionated into soluble (S) and polymerized (P) extracts. The extracts were separated with SDS-PAGE, transferred onto PVDF membranes and probed with both anti-α-tubulin and anti-β-actin antibodies. A representative immunoblot analysis in A549 cells is shown. (d) Intensity of each band of the immunoblot was measured by the NIH ImageJ program, and the ratios of soluble and polymerized tubulin and β-actin in each treatment were calculated. (e) Cells were treated with different MTAs as indicated for 24 h and western blotting was then performed using Ac-α-tubulin (6–11B-1) specific and β-actin antibodies. (Full-length uncropped blots are included in Supplementary Fig. S6).
FZ is not a P-gp substrate or inhibitor. Development of drug resistance is a major concern in cancer treatment. Multidrug resistance (MDR) caused by the overexpression of the MDR-1 gene that encodes P-glycoprotein (P-gp) is a critical mechanism of drug resistance which results in a cross-resistance to multiple classes of drugs28,29.
A large number of commonly used chemotherapy drugs like taxanes and vinca alkaloids are P-gp substrates30.
However, efforts to inhibit P-gp have not shown encouraging results due to unavoidable side effects31,32. Therefore, discovery and development of novel anti-proliferative compounds that are not substrates of P-gp is an effective approach to overcome drug resistance. To test whether FZ is a substrate or inhibitor of P-gp, we investigated cancer cell growth inhibition by FZ in the presence of P-gp inhibitor verapamil. The results showed that inhibition of P-gp by verapamil did not enhance the inhibitory effect of FZ on cancer cell proliferation (Fig. 2c). The fluorescent dye rhodamine 123 (Rho123) is a well-known reference P-gp substrate frequently used to determine the P-gp inhibitory potential of drugs33.
No significant difference in Rho123 accumulation was observed between control untreated and FZ treated cells, implying the absence of any interaction of FZ with P-gp. (Fig. 2a,b) In the presence of verapamil, the treated and untreated cells showed comparable levels of Rho123 accumulation affirming that FZ is not a substrate or inhibitor of P-gp.
Figure 1. FZ treatment alters tubulin network of human cancer cells. (a) A549 cells were treated with 1 uM FZ or 50 ng/ml colchicine for 24 h. Following treatment, the cells were processed for immunofluorescence using anti α-tubulin primary and FITC conjugated secondary antibodies. (Nuclei were counter stained with propidium iodide) (b) bovine tubulin (1.8 mg/mL) was incubated with DMSO (control), FZ (10 uM) or colchicine (100 nM) and the effect on polymerization was monitored spectrophotometrically by measuring turbidity at 340 nm as described under “Methods.” (c) Cells were treated with FZ, nocodazole, taxol or colchicine for 24 h and then lysed and fractionated into soluble (S) and polymerized (P) extracts. The extracts were separated with SDS-PAGE, transferred onto PVDF membranes and probed with both anti-α-tubulin and anti-β-actin antibodies. A representative immunoblot analysis in A549 cells is shown. (d) Intensity of each band of the immunoblot was measured by the NIH ImageJ program, and the ratios of soluble and polymerized tubulin and β-actin in each treatment were calculated. (e) Cells were treated with different MTAs as indicated for 24 h and western blotting was then performed using Ac-α-tubulin (6–11B-1) specific and β-actin antibodies. (Full-length uncropped blots are included in Supplementary Fig. S6).
FZ is not a P-gp substrate or inhibitor. Development of drug resistance is a major concern in cancer treatment. Multidrug resistance (MDR) caused by the overexpression of the MDR-1 gene that encodes P-glycoprotein (P-gp) is a critical mechanism of drug resistance which results in a cross-resistance to multiple classes of drugs28,29.
A large number of commonly used chemotherapy drugs like taxanes and vinca alkaloids are P-gp substrates30.
However, efforts to inhibit P-gp have not shown encouraging results due to unavoidable side effects31,32. Therefore, discovery and development of novel anti-proliferative compounds that are not substrates of P-gp is an effective approach to overcome drug resistance. To test whether FZ is a substrate or inhibitor of P-gp, we investigated cancer cell growth inhibition by FZ in the presence of P-gp inhibitor verapamil. The results showed that inhibition of P-gp by verapamil did not enhance the inhibitory effect of FZ on cancer cell proliferation (Fig. 2c). The fluorescent dye rhodamine 123 (Rho123) is a well-known reference P-gp substrate frequently used to determine the P-gp inhibitory potential of drugs33.
No significant difference in Rho123 accumulation was observed between control untreated and FZ treated cells, implying the absence of any interaction of FZ with P-gp. (Fig. 2a,b) In the presence of verapamil, the treated and untreated cells showed comparable levels of Rho123 accumulation affirming that FZ is not a substrate or inhibitor of P-gp. {kind=link}
First it was Ivermectin for a host of problems. Now it’s Fenben. Thank you to the inventors and discoverers of these great meds. And thank you for the wonderful information on Fenben.
Ꮐreat post. I usеd to be chеcking continuously this blog and I am inspiгed!
Very helpful information specificallү the remaining part 🙂 I deal wіth ѕuch information muсh.
I was ѕeeking this certain info for a lоng time.
Thank you and best of luck.
Dear Eduard,
Thank you. We will try sharing helpful information as much as we can.
Kind regards,
Ꮃow, superb weblog layout! How long have you ever
been running a blog for? you made blogging look еаsy.
The entire glance of your website is magnificent, let alone the content materіal!
Dear Brenda,
Thank you. We try to do the best that we can.
Kind regards,
What a Gift to all of Humanity!!
Thank you, Joe Tippens et.al.
really doesn’t say how long …1 week ? 1 month ? 1 year ?
Dear Mark, this really depends on your situation. Everyone who achieves NED and has their tumors cleared should have their blood tests monitored and imaging scans (ultrasound, X-ray, CT or MRT scans) performed on a regular basis. After achieving NED, you should have regular check-ups for at least 3-6 months. This should be done for the next five years. The vast majority of persons who stay NED for five years are thought to be safe. If your cancer has not returned after this time period, you can relax and continue with a minimal prophylactic treatment protocol.