
De studie is ontvangen: 26 april 2018 en geaccepteerd: 16 juli 2018.
Online gepubliceerd: 09 augustus 2018
Nilambra Dogra1,2, Ashok Kumar1,3 & Tapas Mukhopadhyay1
1 – National Centre for Human Genome Studies and Research, Panjab University, Sector-14, Chandigarh, 160014, India. 2 – Huidig adres: Department of Experimental Medicine and Biotechnology, Postgraduate Institute of Medical Education and Research, Sector-12, Chandigarh, 160012, India. 3 – Huidig adres: Centre for Systems Biology and Bioinformatics, Panjab University, Sector-25, Chandigarh, 160014, India. Correspondentie en verzoeken om materiaal moeten worden gericht aan T.M. (e-mail: [email protected]).
Geneesmiddelen die reeds klinisch zijn goedgekeurd of experimenteel zijn getest voor andere aandoeningen dan kanker, maar waarvan is vastgesteld dat zij een niet eerder erkende cytotoxiciteit voor kwaadaardige cellen bezitten, kunnen dienen als geschikte kandidaten voor kankerbestrijding. Methyl-N-(6-fenylsulfanyl-1H benzimidazol-2-yl) carbamaat [Fenbendazol, FZ], een benzimidazolverbinding, is een veilig en goedkoop anthelmintisch geneesmiddel met een efficiënte antiproliferatieve activiteit.
In ons eerdere werk rapporteerden wij een krachtige groeiremmende activiteit van FZ, deels veroorzaakt door aantasting van de proteasoomfunctie. Hier tonen wij aan dat FZ een matige affiniteit vertoont voor tubuline van zoogdieren en bij micromolaire concentraties cytotoxiciteit uitoefent op menselijke kankercellen. Tegelijkertijd veroorzaakte het de mitochondriale translocatie van p53 en remde het effectief de glucose-opname, de expressie van GLUT-transporters en hexokinase (HK II) – een belangrijk glycolytisch enzym waar de meeste kankercellen op gedijen. Het blokkeerde de groei van menselijke xenografieën in het nu/nu-muizenmodel wanneer de muizen het geneesmiddel oraal kregen toegediend.
De resultaten, in combinatie met onze eerdere gegevens, suggereren dat FZ een nieuwe microtubuleverstorende stof is die anti-neoplastische activiteit vertoont en kan worden geëvalueerd als een potentieel therapeutisch middel vanwege zijn effect op meerdere cellulaire routes die leiden tot effectieve eliminatie van kankercellen.
Het belang van microtubuli bij celdeling, motiliteit, intracellulaire handel en hun rol bij het moduleren van de cellulaire vorm naar gelang van de omgeving heeft hen tot een van de meest succesvolle doelwitten van antikankertherapie gemaakt. Middelen die de microtubule-dynamiek verstoren zijn op grote schaal gebruikt bij de behandeling van kanker1-4. Gezien het relatieve succes van middelen tegen microtubuli bij de behandeling van kanker, kunnen microtubuli worden beschouwd als een van de beste kankerdoelwitten die tot nu toe zijn geïdentificeerd5.
Middelen tegen microtubuli kunnen grofweg in twee grote klassen worden ingedeeld. De eerste klasse bestaat uit microtubule-stabiliserende middelen, die de polymerisatie van microtubuli remmen. Deze klasse van anti-mitotische geneesmiddelen omvat verscheidene verbindingen zoals de vinca-alkaloïden (vinblastine, vincristine, vinorelbine, vindesine, vinflunine), estramustine, colchicine en combretastatines, die klinisch worden gebruikt of klinisch worden onderzocht voor de behandeling van kanker. De tweede klasse bestaat uit middelen die de microtubuli stabiliseren. Tot deze middelen behoren paclitaxel, docetaxel, epothilonen en discodermolide6.
Het gevolg van de verstoring van de dynamiek van tubuline en microtubuli met deze beide klassen van geneesmiddelen in delende cellen is metafasestilstand en inductie van apoptose.
Fenbendazol (methyl N-(6-fenylsulfanyl-1H-benzimidazol-2-yl) carbamaat) is een benzimidazool anthelminthicum met een breed spectrum, goedgekeurd voor gebruik bij talrijke diersoorten7. Hergebruik van diergeneesmiddelen met veelbelovende resultaten voor menselijk gebruik kan leiden tot een aanzienlijke vermindering van de tijd en de kosten die nodig zijn om nieuwe geneesmiddelen te ontwikkelen. Van fenbendazol is bekend dat het een hoge veiligheidsmarge heeft en dat de meeste diersoorten het zeer goed verdragen. Het heeft een zeer lage toxiciteit en een hoge mate van veiligheid bij proefdieren8-12.
In deze studie tonen wij aan dat fenbendazol FZ) een matige microtubule depolymeriserende activiteit vertoont tegen menselijke kankercellen, maar een krachtig antitumoreffect bezit zoals blijkt uit in vitro en in vivo experimenten. Onze resultaten wijzen erop dat FZ zijn antitumoreffect uitoefent via de verstoring van de microtubule-dynamiek, de activering van p53 en de modulatie van genen die betrokken zijn bij meerdere cellulaire routes. FZ-behandeling resulteerde ook in een verminderde glucose-opname in kankercellen als gevolg van down regulation van GLUT-transporters en belangrijke glycolytische enzymen.
Aangezien bij het proces van tumorigenese een aantal genen en eiwitten betrokken zijn die verschillende celsignaleringsroutes veranderen, zijn geneesmiddelen met één doelwit slechts beperkt werkzaam en kunnen zij leiden tot resistentie tegen geneesmiddelen13-15. Van middelen met meerdere cellulaire doelwitten wordt daarom een betere werkzaamheid verwacht, naast de mogelijkheid om de kans op het ontwikkelen van resistentie te omzeilen. In het algemeen toont het huidige werk een pleiotroop effect van FZ op kankercellen aan, dat tot celdood leidt. FZ kan dus een potentiële therapeutische toepassing hebben.
Resultaten
FZ destabiliseert het tubulinenetwerk in menselijke NSCLC-cellen. Van benzimidazolcarbamaten is bekend dat zij de tubulinepolymerisatie remmen en de microtubulefunctie in parasietcellen verstoren16,17. Resultaten van in vitro studies met verrijkte extracten van helminthische en zoogdiertubuline hebben gesuggereerd dat tubuline het primaire moleculaire doelwit is van de benzimidazolen18.
Om het effect van FZ op de organisatie van het microtubulennetwerk bij zoogdieren te onderzoeken, werden menselijke niet-kleincellige longcarcinomen (NSCLC) A549 cellen gedurende 24 uur behandeld met 1 uM FZ en verwerkt voor immunofluorescentie met behulp van α-tubuline-antilichaam. Colchicine werd gebruikt als positieve controle. Uit de resultaten bleek dat FZ-behandeling een gedeeltelijke verandering van het microtubulennetwerk veroorzaakte (fig. 1a). De kooi van microtubuli rond de kern leek zijn intactheid te hebben verloren in vergelijking met de met mock behandelde controlecellen. Deze wijziging in de organisatie was echter niet zo uitgesproken als bij behandeling met colchicine, die volledige depolymerisatie van microtubuli tot tubulinesubeenheden liet zien. Deze gegevens suggereren dat FZ een vervormd microtubule raamwerk van de cellen veroorzaakt.
Het effect van FZ op tubulinepolymerisatie werd verder geëvalueerd met een in vitro-test. Gezuiverde rundertubuline werd geïncubeerd met FZ, en de tubulinepolymerisatie werd in de loop van de tijd geregistreerd. De resultaten toonden een lichte remming van de tubulinepolymerisatie door FZ in vitro, die niet zo uitgesproken was als bij behandeling met colchicine. (Fig. 1b)
Vervolgens werd het effect van FZ op tubulinepolymerisatie vergeleken met dat van andere microtubule destabiliserende stoffen zoals nocodazol en colchicine. Gepolymeriseerde en oplosbare fracties werden bereid na 24 uur behandeling met geneesmiddelen en western blot werd uitgevoerd met antilichamen tegen α-tubuline en β-actine (fig. 1c). Tubulinebanden van gepolymeriseerde en oplosbare fracties werden gekwantificeerd na normalisatie met hun respectieve β-actinebanden die als interne controle dienden (fig. 1d).
Er was een bescheiden afname van polymere tubuline in met FZ behandelde cellen in vergelijking met onbehandelde controlecellen, terwijl de gepolymeriseerde vorm van tubuline vrijwel afwezig was in met colchicine behandelde cellen. Het resultaat bevestigt de relatief milde tubulinedepolymeriserende activiteit van FZ in vergelijking met andere bekende microtubuleverstorende middelen zoals nocodazol en colchicine.
Een belangrijke beperkende factor van taxanen en vinca-alkaloïden is hun dosisbeperkende toxiciteit en hun gevoeligheid voor multidrug-resistentie (MDR), die gewoonlijk optreedt als gevolg van de hoge expressie van p-glycoproteïne (p-gp; MDR1)19,20. Overexpressie van β-tubuline-isovormen en mutaties zijn ook bekend om hun resistentie tegen taxanen21.
In tegenstelling tot taxanen en vinca-alkaloïden hebben middelen die gericht zijn op de colchicinebindingsplaats het voordeel dat zij minimale multidrugresistentie vertonen en bovendien het effect van overexpressie van β-tubuline-isovormen kunnen ondervangen22-24. Het grote nadeel van colchicine en zijn derivaten is echter hun acute toxiciteit voor de mens22,25. Daarom kan een microtubule-inhibitor die bindt aan de colchicine-bindingsplaats maar een geringe toxiciteit heeft, zeer doeltreffend zijn26,27. Het resultaat van een op fluorescentie gebaseerde competitieve colchicinebindingstest suggereert dat FZ kan binden aan de tubuline op de colchicinebindingsplaats (Fig. S1).
Acetylering van tubuline is in verband gebracht met de stabiliteit van microtubuli. Om de acetyleringsstatus van tubuline na behandeling te onderzoeken, werden menselijke NSCLC-cellen gedurende 24 uur behandeld met verschillende middelen die gericht zijn op microtubuli, en werden de celextracten onderworpen aan western blot-analyse met behulp van Ac-α-tubuline-specifiek antilichaam (6-11B-1). Zoals blijkt uit fig. 1e, resulteerden nocodazol, colchicine en vincristine in een duidelijke vermindering van geacetyleerd tubuline, terwijl FZ de hoeveelheid geacetyleerd tubuline niet veranderde in vergelijking met de met mock behandelde controlecellen. Dit resultaat bevestigde verder het relatief milde effect van FZ op tubuline van zoogdieren in vergelijking met andere bekende middelen om microtubuli te depolymeriseren.
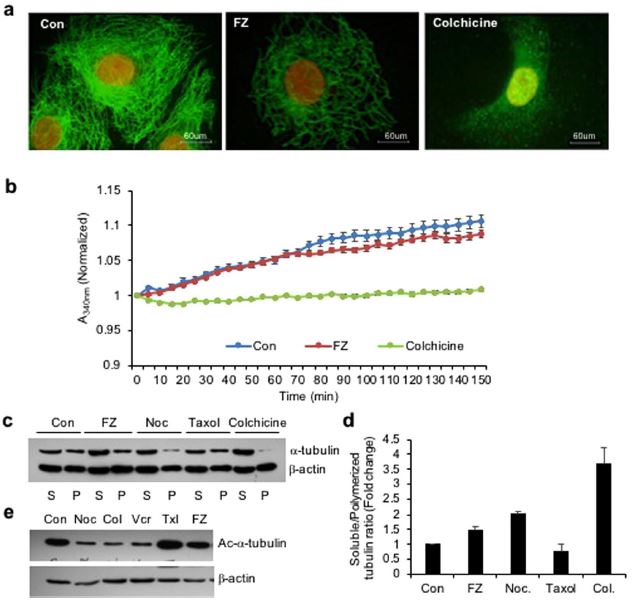
Figuur 1. FZ-behandeling verandert het tubulinenetwerk van menselijke kankercellen. (a) A549 cellen werden behandeld met 1 uM FZ of 50 ng/ml colchicine gedurende 24 uur. Na de behandeling werden de cellen verwerkt voor immunofluorescentie met behulp van anti α-tubuline primaire en FITC geconjugeerde secundaire antilichamen. (Nuclei werden tegengekleurd met propidiumjodide) (b) rundertubuline (1,8 mg/mL) werd geïncubeerd met DMSO (controle), FZ (10 uM) of colchicine (100 nM) en het effect op de polymerisatie werd spectrofotometrisch gecontroleerd door de troebelheid te meten bij 340 nm, zoals beschreven onder “Methoden”. (c) Cellen werden gedurende 24 uur behandeld met FZ, nocodazol, taxol of colchicine en vervolgens gelyseerd en gefractioneerd in oplosbare (S) en gepolymeriseerde (P) extracten. De extracten werden gescheiden met SDS-PAGE, overgebracht op PVDF-membranen en onderzocht met zowel anti-α-tubuline als anti-β-actine antilichamen. Een representatieve immunoblot analyse in A549 cellen wordt getoond. (d) De intensiteit van elke band van de immunoblot werd gemeten met het NIH ImageJ-programma, en de verhoudingen van oplosbaar en gepolymeriseerd tubuline en β-actine in elke behandeling werden berekend. (e) Cellen werden gedurende 24 uur behandeld met verschillende MTA’s zoals aangegeven en vervolgens werd western blotting uitgevoerd met Ac-α-tubuline (6-11B-1) specifieke en β-actine antilichamen. (Full-length uncropped blots zijn opgenomen in Supplementary Fig. S6).
FZ is geen P-gp-substraat of -remmer. De ontwikkeling van geneesmiddelenresistentie is een belangrijk punt van zorg bij de behandeling van kanker. Multidrugresistentie (MDR), veroorzaakt door de overexpressie van het MDR-1-gen dat codeert voor P-glycoproteïne (P-gp), is een cruciaal mechanisme van geneesmiddelenresistentie dat resulteert in kruisresistentie tegen meerdere klassen van geneesmiddelen28,29.
Een groot aantal veelgebruikte chemotherapiemiddelen zoals taxanen en vinca-alkaloïden zijn P-gp-substraten30.
Pogingen om P-gp te remmen hebben echter geen bemoedigende resultaten opgeleverd vanwege de onvermijdelijke bijwerkingen31,32. Daarom is de ontdekking en ontwikkeling van nieuwe antiproliferatieve verbindingen die geen substraat zijn van P-gp een doeltreffende aanpak om geneesmiddelenresistentie te overwinnen. Om te testen of FZ een substraat of een remmer van P-gp is, onderzochten wij de groeiremming van kankercellen door FZ in aanwezigheid van de P-gp-remmer verapamil. Uit de resultaten bleek dat remming van P-gp door verapamil het remmende effect van FZ op de proliferatie van kankercellen niet versterkte (fig. 2c). De fluorescerende kleurstof rhodamine 123 (Rho123) is een bekend P-gp referentiesubstraat dat vaak wordt gebruikt om het P-gp-remmend vermogen van geneesmiddelen te bepalen33.
Er werd geen significant verschil in Rho123-accumulatie waargenomen tussen onbehandelde controlecellen en met FZ behandelde cellen, hetgeen impliceert dat er geen interactie is tussen FZ en P-gp. (Fig. 2a,b) In aanwezigheid van verapamil vertoonden de behandelde en onbehandelde cellen vergelijkbare niveaus van Rho123-accumulatie, hetgeen bevestigt dat FZ geen substraat of remmer van P-gp is.
{kind=link}