
L’étude a été reçue : 26 avril 2018 et acceptée : 16 juillet 2018.
Publiée en ligne : 09 août 2018
Nilambra Dogra1,2, Ashok Kumar1,3 & Tapas Mukhopadhyay1
1 – Centre national d’études et de recherches sur le génome humain, Université Panjab, Sector-14, Chandigarh, 160014, Inde. 2 – Adresse actuelle : Département de médecine expérimentale et de biotechnologie, Postgraduate Institute of Medical Education and Research, Sector-12, Chandigarh, 160012, Inde. 3 – Adresse actuelle : Centre for Systems Biology and Bioinformatics, Panjab University, Sector-25, Chandigarh, 160014, Inde. La correspondance et les demandes de matériel doivent être adressées à T.M. (courriel : [email protected]).
Les médicaments déjà approuvés cliniquement ou testés expérimentalement pour des affections autres que le cancer, mais dont la cytotoxicité à l’égard des cellules malignes est inconnue jusqu’à présent, peuvent constituer des candidats anticancéreux appropriés. Le méthyl N-(6-phénylsulfanyl-1H benzimidazol-2-yl) carbamate [Fenbendazole, FZ], un composé benzimidazole, est un médicament anthelminthique sûr et peu coûteux qui possède une activité anti-proliférative efficace.
Dans nos travaux antérieurs, nous avons signalé une puissante activité d’inhibition de la croissance du FZ causée en partie par une altération de la fonction protéasomale. Nous montrons ici que le FZ présente une affinité modérée pour la tubuline des mammifères et qu’il exerce une cytotoxicité sur les cellules cancéreuses humaines à des concentrations micromolaires. Simultanément, il provoque la translocation mitochondriale de p53 et inhibe efficacement l’absorption du glucose, l’expression des transporteurs GLUT ainsi que l’hexokinase (HK II) – une enzyme glycolytique clé dont se nourrissent la plupart des cellules cancéreuses. Il a bloqué la croissance des xénogreffes humaines dans le modèle de souris nu/nu lorsque les souris ont été nourries avec le médicament par voie orale.
Ces résultats, associés à nos données antérieures, suggèrent que le FZ est un nouvel agent interférant avec les microtubules qui présente une activité anti-néoplasique et peut être évalué en tant qu’agent thérapeutique potentiel en raison de son effet sur de multiples voies cellulaires conduisant à l’élimination efficace des cellules cancéreuses.
L’importance des microtubules dans la division cellulaire, la motilité, le trafic intracellulaire et leur rôle dans la modulation de la forme cellulaire en fonction de l’environnement en ont fait l’une des cibles les plus efficaces de la thérapie anticancéreuse. Les agents qui perturbent la dynamique des microtubules ont été largement utilisés dans le traitement du cancer1-4. Compte tenu du succès relatif des agents mitotiques dans le traitement du cancer, les microtubules peuvent être considérés comme l’une des meilleures cibles anticancéreuses identifiées à ce jour5.
Les agents ciblant les microtubules peuvent être classés en deux grandes catégories. La première comprend les agents déstabilisant les microtubules, qui inhibent la polymérisation des microtubules. Cette classe de médicaments antimitotiques comprend plusieurs composés tels que les alcaloïdes de la pervenche (vinblastine, vincristine, vinorelbine, vindésine, vinflunine), l’estramustine, la colchicine et les combretastatines, qui sont utilisés cliniquement ou font l’objet d’études cliniques pour le traitement du cancer. La deuxième classe comprend les agents stabilisant les microtubules. Ces agents comprennent le paclitaxel, le docétaxel, les épothilones et le discodermolide6.
La perturbation de la dynamique de la tubuline et des microtubules par ces deux classes de médicaments dans les cellules en division a pour conséquence l’arrêt de la métaphase et l’induction de l’apoptose.
Le fenbendazole (N-(6-phénylsulfanyl-1H-benzimidazol-2-yl) carbamate de méthyle) est un anthelminthique benzimidazole à large spectre dont l’utilisation est approuvée chez de nombreuses espèces animales7. La réaffectation à l’usage humain de médicaments vétérinaires dont les résultats sont prometteurs peut permettre de réduire considérablement le temps et les coûts nécessaires au développement de nouveaux médicaments. Le fenbendazole est connu pour avoir une marge de sécurité élevée et la plupart des espèces le tolèrent très bien. Il présente un degré de toxicité très faible et un degré de sécurité élevé chez les animaux de laboratoire8-12.
Dans cette étude, nous montrons que le fenbendazole (FZ) présente une activité modérée de dépolymérisation des microtubules sur les cellules cancéreuses humaines, mais qu’il possède un puissant effet antitumoral, comme le montrent les expériences in vitro et in vivo. Nos résultats indiquent que le FZ exerce son effet antitumoral par la perturbation de la dynamique des microtubules, l’activation de p53 et la modulation de gènes impliqués dans de multiples voies cellulaires. Le traitement par FZ a également entraîné une réduction de l’absorption du glucose dans les cellules cancéreuses en raison de la régulation des transporteurs GLUT et des enzymes glycolytiques clés.
Étant donné que le processus de tumorigenèse implique un certain nombre de gènes et de protéines modifiant diverses voies de signalisation cellulaire, les médicaments à cible unique présentent une efficacité limitée et peuvent entraîner une résistance aux médicaments13-15. Les agents ayant des cibles cellulaires multiples devraient donc être plus efficaces et permettre de contourner le risque de développement d’une résistance. Dans l’ensemble, le présent travail démontre un effet pléiotropique du FZ sur les cellules cancéreuses conduisant à la mort cellulaire. Le FZ pourrait donc avoir une application thérapeutique potentielle.
Résultats
Le FZ déstabilise le réseau de tubuline dans les cellules humaines NSCLC. Les carbamates benzimidazoles ont été signalés comme inhibant la polymérisation de la tubuline et perturbant la fonction des microtubules dans les cellules parasitaires16,17. Les résultats d’études in vitro utilisant des extraits enrichis de tubuline helminthique et mammalienne ont suggéré que la tubuline est la principale cible moléculaire des benzimidazoles18.
Par conséquent, pour examiner l’effet du FZ sur l’organisation du réseau de microtubules chez les mammifères, les cellules A549 du carcinome pulmonaire non à petites cellules (NSCLC) humain ont été traitées avec 1 uM de FZ pendant 24 h et traitées par immunofluorescence à l’aide de l’anticorps α-tubuline. La colchicine a été utilisée comme contrôle positif. Les résultats ont montré que le traitement par FZ provoquait une altération partielle du réseau de microtubules (Fig. 1a). La cage de microtubules autour du noyau semble avoir perdu son intégrité par rapport aux cellules témoins traitées par simulacre. Cependant, cette modification de l’organisation n’était pas aussi marquée que dans le cas du traitement à la colchicine, qui a montré une dépolymérisation complète des microtubules en sous-unités de tubuline. Ces données suggèrent que le FZ provoque une distorsion de la structure des microtubules dans les cellules.
L’effet du FZ sur la polymérisation de la tubuline a été évalué de manière plus approfondie par un test in vitro. De la tubuline bovine purifiée a été incubée avec du FZ et la polymérisation de la tubuline a été enregistrée au cours du temps. Les résultats ont montré une légère inhibition de la polymérisation de la tubuline par le FZ in vitro, qui n’était pas aussi prononcée que dans le cas d’un traitement à la colchicine. (Fig. 1b)
Ensuite, l’effet du FZ sur la polymérisation de la tubuline a été comparé à celui d’autres agents déstabilisateurs de microtubules comme le nocodazole et la colchicine. Des fractions polymérisées et solubles ont été préparées après 24 heures de traitement médicamenteux et un western blot a été réalisé en utilisant les anticorps α-tubuline et β-actine (Fig. 1c). Les bandes de tubuline des fractions polymérisées et solubles ont été quantifiées après normalisation avec leurs bandes de β-actine respectives qui ont servi de contrôle interne (Fig. 1d).
Une diminution modeste de la tubuline polymérique a été observée dans les cellules traitées par FZ par rapport aux cellules témoins non traitées, alors que la forme polymérisée de la tubuline était presque absente dans les cellules traitées par la colchicine. Ce résultat confirme l’activité dépolymérisante de la tubuline relativement faible du FZ par rapport à d’autres agents perturbateurs des microtubules connus comme le nocodazole et la colchicine.
L’un des principaux facteurs limitant les taxanes et les vinca-alcaloïdes est leur toxicité à dose limitée et leur susceptibilité à la résistance aux médicaments multiples (MDR), généralement due à l’expression élevée de la p-glycoprotéine (p-gp ; MDR1)19,20. La surexpression des isoformes de β-tubuline et les mutations sont également connues pour conférer une résistance aux taxanes21.
Contrairement aux taxanes et aux alcaloïdes de la pervenche, les agents ciblant le site de liaison de la colchicine présentent l’avantage de présenter une multirésistance minimale aux médicaments, en plus de leur capacité à surmonter l’effet de la surexpression des isoformes de la β-tubuline22-24. Cependant, le principal inconvénient de la colchicine et de ses dérivés est leur toxicité aiguë pour l’homme22,25. Par conséquent, un inhibiteur de microtubules qui se lie au site de liaison de la colchicine tout en présentant une faible toxicité peut s’avérer très efficace26,27. Les résultats d’un essai de liaison compétitive à la colchicine basé sur la fluorescence suggèrent que le FZ peut se lier à la tubuline au niveau du site de liaison de la colchicine (Fig. S1).
L’acétylation de la tubuline a été associée à la stabilité des microtubules. Par conséquent, pour examiner l’état d’acétylation de la tubuline après traitement, des cellules humaines NSCLC ont été traitées avec différents agents ciblant les microtubules pendant 24 heures et les extraits cellulaires ont été soumis à une analyse par western blot en utilisant l’anticorps spécifique Ac-α-tubuline (6-11B-1). Comme le montre la figure 1e, alors que le nocodazole, la colchicine et la vincristine ont entraîné une réduction marquée de la tubuline acétylée, le FZ n’a pas modifié la quantité de tubuline acétylée par rapport aux cellules témoins traitées à l’aide d’un simulacre. Ce résultat confirme l’effet relativement faible du FZ sur la tubuline des mammifères par rapport à d’autres agents connus de dépolymérisation des microtubules.
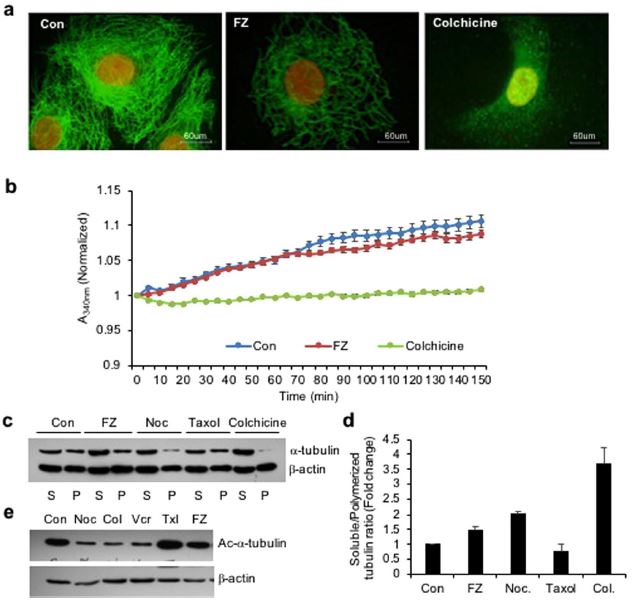
Figure 1. Le traitement par FZ modifie le réseau de tubuline des cellules cancéreuses humaines. (a) Les cellules A549 ont été traitées avec 1 uM de FZ ou 50 ng/ml de colchicine pendant 24 heures. Après le traitement, les cellules ont été traitées pour l’immunofluorescence en utilisant des anticorps primaires anti-α-tubuline et des anticorps secondaires conjugués au FITC. (Les noyaux ont été contre-colorés avec de l’iodure de propidium). (b) La tubuline bovine (1,8 mg/mL) a été incubée avec du DMSO (contrôle), du FZ (10 uM) ou de la colchicine (100 nM) et l’effet sur la polymérisation a été contrôlé par spectrophotométrie en mesurant la turbidité à 340 nm, comme décrit dans la section « Méthodes ». (c) Les cellules ont été traitées avec FZ, nocodazole, taxol ou colchicine pendant 24 heures, puis lysées et fractionnées en extraits solubles (S) et polymérisés (P). Les extraits ont été séparés par SDS-PAGE, transférés sur des membranes PVDF et analysés avec des anticorps anti-α-tubuline et anti-β-actine. Une analyse immunoblot représentative des cellules A549 est présentée. (d) L’intensité de chaque bande de l’immunoblot a été mesurée par le programme NIH ImageJ, et les ratios de tubuline soluble et polymérisée et de β-actine dans chaque traitement ont été calculés. (e) Les cellules ont été traitées avec différents MTA comme indiqué pendant 24 h et un western blotting a ensuite été réalisé en utilisant des anticorps spécifiques Ac-α-tubuline (6-11B-1) et β-actine. (Les blots non recoupés de pleine longueur sont inclus dans la Fig. supplémentaire S6).
Le FZ n’est pas un substrat ou un inhibiteur de la P-gp. Le développement de la résistance aux médicaments est une préoccupation majeure dans le traitement du cancer. La multirésistance aux médicaments (MDR) causée par la surexpression du gène MDR-1 qui code pour la P-glycoprotéine (P-gp) est un mécanisme critique de résistance aux médicaments qui entraîne une résistance croisée à plusieurs classes de médicaments28,29.
Un grand nombre de médicaments de chimiothérapie couramment utilisés, comme les taxanes et les alcaloïdes de la pervenche, sont des substrats de la P-gp30.
Toutefois, les efforts déployés pour inhiber la P-gp n’ont pas donné de résultats encourageants en raison d’effets secondaires inévitables31,32. Par conséquent, la découverte et le développement de nouveaux composés anti-prolifératifs qui ne sont pas des substrats de la P-gp constituent une approche efficace pour surmonter la résistance aux médicaments. Pour vérifier si le FZ est un substrat ou un inhibiteur de la P-gp, nous avons étudié l’inhibition de la croissance des cellules cancéreuses par le FZ en présence de vérapamil, un inhibiteur de la P-gp. Les résultats ont montré que l’inhibition de la P-gp par le vérapamil ne renforçait pas l’effet inhibiteur du FZ sur la prolifération des cellules cancéreuses (Fig. 2c). Le colorant fluorescent rhodamine 123 (Rho123) est un substrat de référence bien connu de la P-gp, fréquemment utilisé pour déterminer le potentiel d’inhibition de la P-gp par les médicaments33.
Aucune différence significative dans l’accumulation de Rho123 n’a été observée entre les cellules témoins non traitées et les cellules traitées au FZ, ce qui implique l’absence d’interaction du FZ avec la P-gp. (Fig. 2a,b) En présence de vérapamil, les cellules traitées et non traitées ont montré des niveaux comparables d’accumulation de Rho123, ce qui confirme que le FZ n’est pas un substrat ou un inhibiteur de la P-gp.
{kind=link}