
Cancer is a daunting disease where abnormal cells divide uncontrollably and acquire the ability to spread beyond its site of origin, infiltrating and destroying distant body tissues and organs. This latter process is called metastasis and is the single most common cause of cancer-related death (Figure 1).
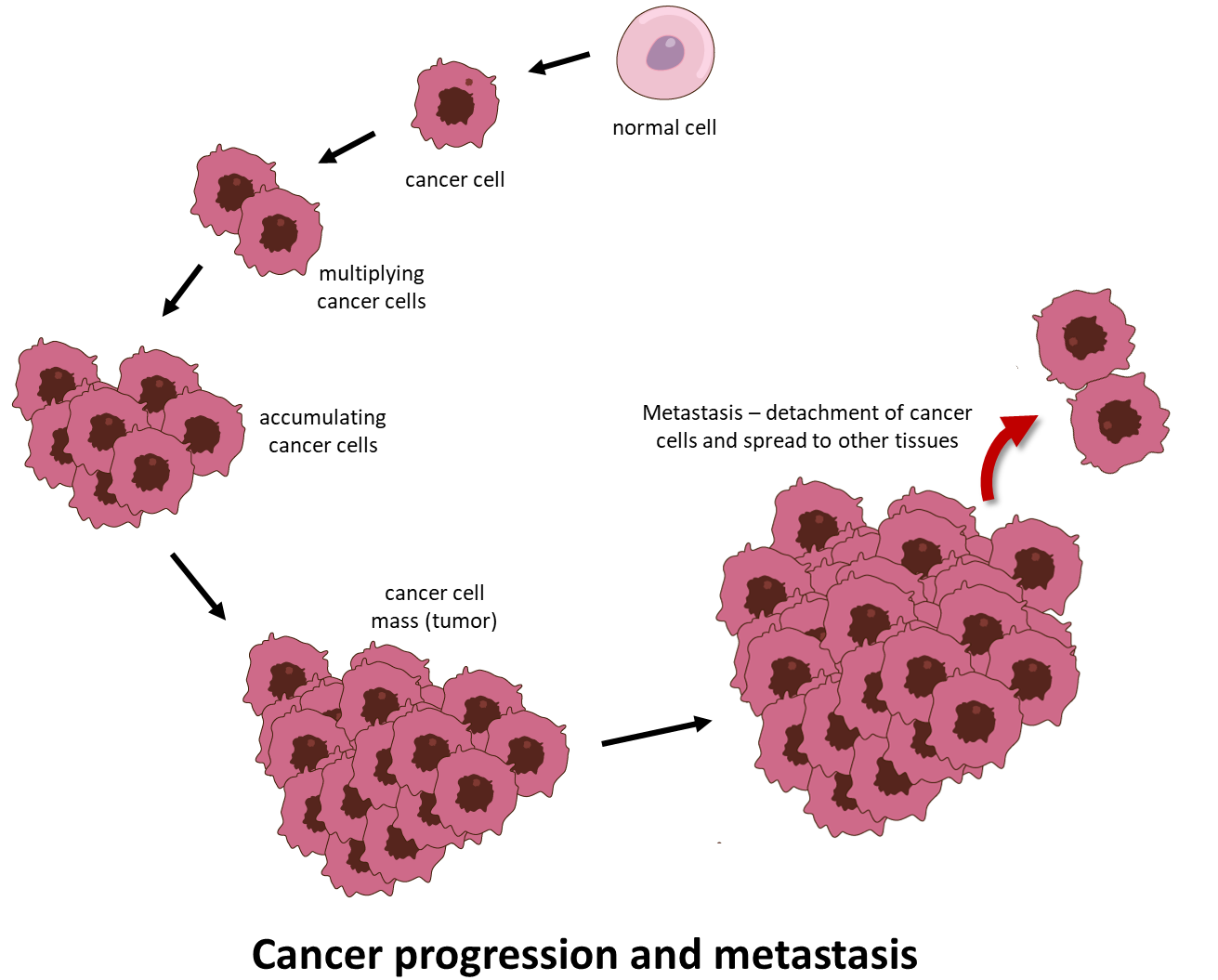
In 2020, 19.3 million new cases of cancer were reported worldwide; out of which a staggering 10.0 million deaths were estimated.1 Cancer incidences continue to increase and so does the incidence of cancer related death. Rapid advances in cancer research over the past five decades have revealed the fact that during tumorigenesis, cancer cells acquire alterations in cell physiology that differentiate them from normal cells. These acquired capabilities, also known as hallmarks of cancer (Figure 2) are shared by all types of cancer.2 These shared traits include:
- Self-sufficiency in growth signals. Unlike normal cells, cancer cells are not dependent on exogenous growth factors for active proliferation. Instead, they acquire the ability to synthesize Growth Factors to which they are responsive, creating a positive-feedback signaling loop.
- Evasion of growth suppressors. Unlike normal cells, wherein multiple anti-proliferative soluble growth inhibitors operate to maintain cellular quiescence and tissue homeostasis, cancer cells somehow learn to evade anti-proliferative signals that slow down cell growth. Cancer cells therefore continue to proliferate even in the presence of growth suppressors.
- Resistance to cell death. Cancer cells oppose natural cell death pathways that eliminate unnecessary or potentially dangerous cells.
- Angiogenesis. Cancer cells rapidly grow blood vessels via angiogenesis to connect to the circulatory system to access oxygen and nutrients to survive and proliferate.
- Invasion and metastasis. Cancer cells transfer from the primary site to distant locations a) to evade immune surveillance, b) to overcome lack of oxygen and necessary nutrients due to large tumor size, c) to escape cell death caused by accumulation of excess lactic acid.
- Immune evasion. Tumors can evade attacks from the immune surveillance system through various mechanisms such as restricting recognition of antigens i.e. it fools the immune system from recognizing rogue cancer cells and also induces exhaustion of T cells, the soldiers of the immune system that recognize rogue cancer cells.
- Tumor-promoting inflammation. In a normal inflammatory response, immune cell produces chemicals that kill a pathogen. These chemicals, known as reactive oxygen species, can also damage the DNA of normal cells, which in turn increases the risk of mutations and subsequently drives cancer development due to chronic inflammation.
- Mutation(s) and Genomic instability. This occurs due to the increased tendency of genome alteration during cell division. Aberrant DNA replication contributes to acquisition of mutation(s) and an unstable genome which in turn leads to development of malignancy.
- Epigenetic reprogramming. These are changes in the underlying chromatin structure of a gene. Such changes can turn on a cancer-causing oncogene or turn off a tumor suppressor gene and assist the process of tumorigenesis.
- Polymorphic genomes. Or genetic variations in genes that regulate DNA mismatch repair, cell cycle regulation, metabolism and immunity increase susceptibility to cancer.
- Phenotypic plasticity. The ability of cancer cells to undergo dynamic changes in the shape, size, morphology etc. helps drive immune evasion and drug resistance and promotes invasion and distant migration of cancer cells.
- Metabolic reprogramming. The ability of cancer cells to alter their metabolism to meet the increased energy demand of rapidly proliferating cancer cells, and in doing so to become ‘immortal.’
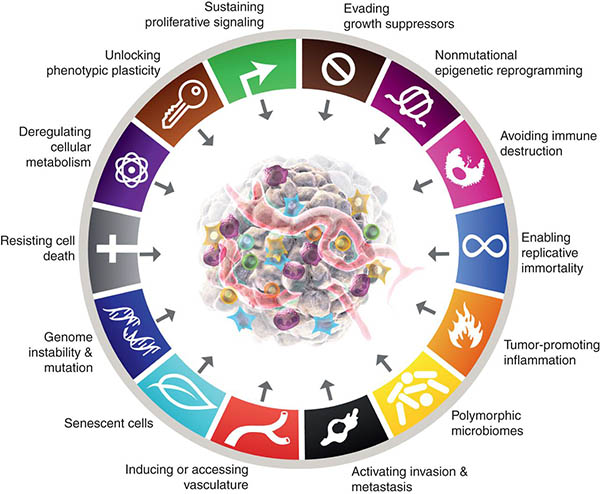
(Image Courtesy of Cancer Discovery Journal https://www.aacr.org/blog/2022/01/21/new-dimensions-in-cancer-biology-updated-hallmarks-of-cancer-published/)
Current Challenges to Find the Right Cancer Treatment
Since there are multiple possible factors which drive transformation of normal cells to become malignant cancer cells, prevention remains very difficult and eradication of cancer once it is already established still poses myriad challenges. Conventional cancer therapy, including chemotherapy and radiation therapy, primarily targets actively proliferating cells.
Chemotherapeutic drugs do not differentiate between actively dividing normal cells and cancer cells. Therefore, severe toxic effects are observed in patients due to inadvertent but unavoidable loss of normal cells. This results in neurotoxicity, cardiotoxicity, gastrointestinal toxicity, and immune suppression. Furthermore, chemotherapeutic drugs are often associated with resistance.3,4
Therefore, many cancer patients whose disease recurs after a brief remission are in dire need of alternative medicines that also address other cancer hallmarks and cause less toxicity.
Metabolic Reprogramming: The Key Reason Why Existing Cancer Therapies Fail
Glucose is the main energy source for normal, eukaryotic, non-cancer cells. During glucose metabolism, normal cells produce CO2 via oxidative phosphorylation under aerobic conditions or lactate via glycolysis under anaerobic conditions. In cancer cells, the metabolic shift towards excessive glucose consumption—over 200 times the normal rate, known as aerobic glycolysis or the Warburg effect—plays a pivotal role.5,6
Unlike in normal tissues, where cells only exhibit glycolysis under oxygen-limited or anerobic conditions, cancer cells reprogram the metabolic pathway to carry out glycolysis even in aerobic conditions to meet the high energy demand of uncontrolled cell division; this is called the Warburg effect or aerobic glycolysis and is a critical trait that distinguishes cancer cells from normal cells.
This metabolic alteration leads to an overproduction of lactate, which is then expelled from the cells. This expulsion leads to lactic acid accumulation, which promotes acidification of the tumor microenvironment, a condition that can suppress the immune system and potentially reduce the efficacy of certain immunotherapy treatments.
Why Are Low Sugar/Ketogenic Diets Ineffective?
Furthermore, strategies like ketogenic diets or sugar avoidance are generally ineffective in this context. These approaches often accelerate cachexia, a severe form of muscle and weight loss, hastening the progression to mortality.7
This ineffectiveness is primarily because cancer cells are adept at extracting essential nutrients from muscles (notably glutamine) and other tissues for their sustenance, thereby undermining the intended impact of dietary restrictions.
Repurposing Non-Cancer Drugs to Fight Cancer
The development of new anti-cancer drugs is becoming increasingly difficult and cost prohibitive. To find an effective therapeutic cure for cancer, drugs that are already clinically approved or experimentally tested for conditions other than cancer, but are found to possess previously unrecognized cytotoxicity towards malignant cells are proving to be very useful.
Cancer researchers have identified certain drug molecules which could serve as fitting anti-cancer candidates alongside conventional therapies. Therefore, repurposing drugs with anti-cancer efficacy can be considered an important strategy for cancer therapy today.8
Because of the above outlined mechanism of metabolic reprogramming by which standard cancer therapies typically fail, it would make the most sense to focus on repurposed drug candidates that combat this very mechanism.
Thus, while many promising drug candidates exist to complement existing anti-cancer therapies, there is strong rationale to focus on those drugs that overcome one specific hallmark of cancer in particular: metabolic reprogramming.
Drugs That Help Overcome Metabolic Reprogramming in Cancer: Fenbendazole, Sodium dichloroacetate (DCA) and 2-deoxy-d-glucose (2-DG)
a) How the 3 drugs have unique, independent and synergistic mechanisms to combat excessive glucose consumption in cancer cells:
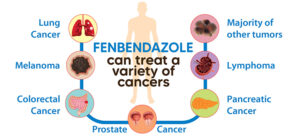
i. Fenbendazole – reduces GLUT transporters and affects hexokinase 2
Fenbendazole (FBZ) (methyl N-(6-phenylsulfanyl-1H-benzimidazol-2-yl) carbamate) is a class of benzimidazole drugs, which finds usage as an anti-cancer drug. FBZ has proven anticancer effect as it arrests cell proliferation, lowers glucose uptake and triggers apoptotic cell death.9 ,10,11
Repurposing of Fenbendazole is hence an important option in cancer therapy that can reduce considerable time and cost required to develop new drugs.
Cancer is associated with a metabolic disorder wherein the cancer cells selectively prefer inefficient aerobic glycolysis (which generates only 2 molecules of ATP) over mitochondrial oxidative phosphorylation (which generates 38 molecules of ATP) to meet their energy needs. Aerobic Glycolysis is an energy inefficient process, so the cancer cells’ need to increase their glucose consumption by several folds.
Cancer cells increase uptake of glucose by upregulating GLUT1, a membrane protein which facilitates basal uptake of glucose. FBZ treatment results in reduced glucose uptake in cancer cells due to downregulation of GLUT transporters and other key glycolytic enzymes like Hexokinase II (HKII).
In order to drive aerobic glycolysis, many glycolytic enzymes are upregulated during tumorigenesis. Hexokinase II (HKII), a key glycolytic enzyme, plays a critical role in glucose retention and metabolism by phosphorylating glucose to glucose-6-phosphate, which can then enter glycolysis.
HKII is thus a highly advantageous enzyme for cancer cell survival and proliferation. FBZ impairs the enzymatic function of HKII in cancer cells leading to reduced glucose retention in FBZ treated cells, eventually the cancer cells die because they cannot utilize glucose as an energy source via aerobic glycolysis.11
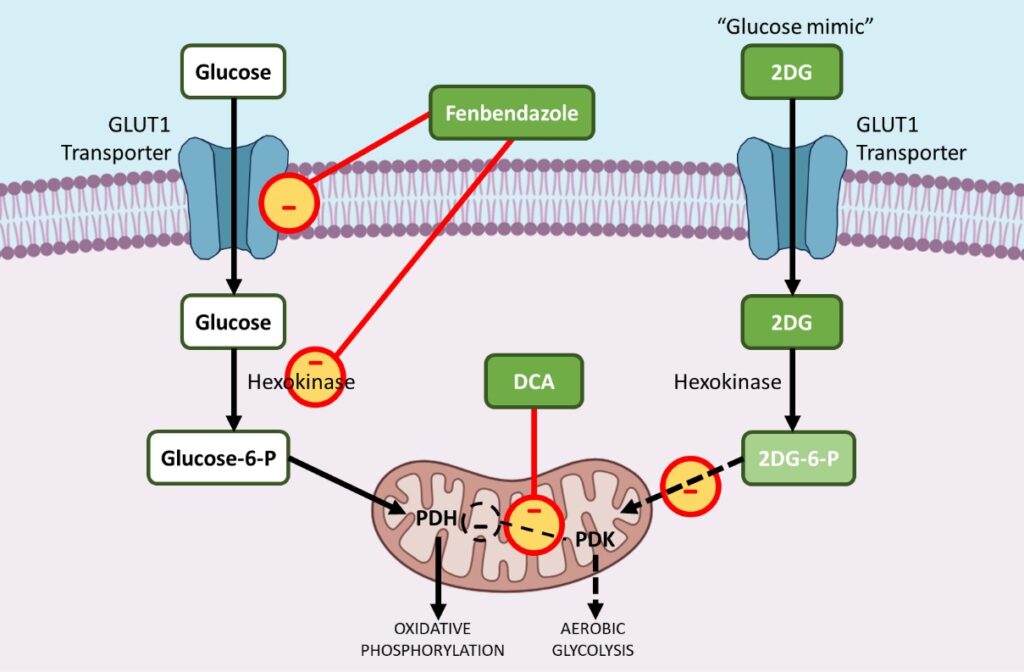
Overcoming Metabolic Programming in Cancer with Fenbendazole, DCA and 2-DG
ii. DCA – inhibits pyruvate dehydrogenase kinase (PDK)
Sodium dichloroacetate (DCA), a small molecule already used to treat acute and chronic lactic acidosis, has been largely repurposed as an anticancer drug.12,13
Cancer cells draw energy from excessive glycolysis instead of mitochondrial oxidative phosphorylation. By upregulating aerobic glycolysis and increasing the acidity of the tumor microenvironment, cancer cells have a clear advantage. Metabolic acidosis in the tumor microenvironment inhibits apoptosis or natural cell death mechanisms and helps build resistance against drugs.
During carcinogenesis most Glycolytic enzymes are upregulated while enzymes that help oxidative phosphorylation in mitochondria are preferentially downregulated. Pyruvate dehydrogenase kinase (PDK) is a gate-keeping enzyme that regulates the flux of glucose into the mitochondria, the seat of oxidative phosphorylation. In the presence of activated PDK, pyruvate dehydrogenase (PDH), a pro-oxidative phosphorylation enzyme is inhibited, limiting the entry of pyruvate into the mitochondria, where glucose oxidation can take place.
This shifts the metabolism towards glycolysis. DCA selectively targets cancer cells’ shifting their metabolism from glycolysis to oxidative phosphorylation by inhibition of pyruvate dehydrogenase kinase (PDK), the inhibitor of pyruvate dehydrogenase (PDH).
The activation of PDH enzymes favors oxidative phosphorylation instead of aerobic glycolysis, the preferred pathway via which cancer cells meet this high energy demand, thus disrupting the metabolic advantage of cancer cells and rendering the cancer cells more sensitive to chemotherapeutic drugs.
iii. 2-DG – a glucose analogue or ‘mimic’ that blocks key enzymes like hexokinase and glucose-6-phosphate isomerase
2-deoxy-D-glucose (2-DG) is a natural, non metabolizable glucose analog, which has the 2-hydroxyl group replaced by hydrogen. 2DG competitively inhibits glucose uptake because glucose and 2-DG are both transported inside the cell by Glucose transporting enzyme (GLUT1). 14,15,16
When less glucose is transported inside the cancer cell, less aerobic glycolysis can occur. Reduction in glycolysis ultimately stops active cell proliferation of the cancer cells.
Secondly, after entering the cell, hexokinase phosphorylates both glucose and 2DG to form glucose-6-phosphate and 2-Deoxy-D-glucose-6-phosphate (2DG-6-P) respectively.
2DG-6-P cannot be further metabolized via glycolysis. As such; it acts to competitively inhibit the production of glucose-6-phosphate from glucose during glycolysis and in doing so inhibits glucose metabolism in cancer cells.
b) How the 3 drugs have unique, independent and synergistic mechanisms to induce selective apoptosis in cancer cells but not in normal cells
i. Fenbendazole – interacts with β-tubulin to prevent polymerization to form microtubules leading to cell cycle arrest and apoptotic cell death
During cell division, microtubules, which are made out of tubulin subunits, form spindles to enable correct chromosomal segregation. FBZ disrupts tubulin polymerization, an essential step during cell proliferation, ensuring cell cycle arrest in actively dividing cancer cells.
Whether it be inhibiting increased uptake of glucose or targeting microtubules to arrest uncontrolled cell division, FBZ triggers cancer cell apoptosis as a common end result.
ii. DCA – increases reactive oxygen species (ROS) in cancer cells, oxidative stress, caspase activation and apoptosis
Dichloroacetate (DCA) is an inhibitor of the pyruvate dehydrogenase kinase (PDK) which phosphorylates PDH to phospho PDH, the active substrate in aerobic glycolysis.13
The inhibition of PDK and activation of PDH are reported to be associated with enhanced production of reactive oxygen species (ROS). ROS induce double-strand breaks in the DNA of cancer cells and enhance their sensitivity to radiation therapy and apoptosis.
iii. 2-DG – deprives cancer cells of nutrients, causes oxidative stress and apoptotic cell death
Since 2DG mimics glucose deprivation conditions, it inhibits aerobic glycolysis, which ultimately leads to deprivation of nutrients and cell death.
Further, GFAT1 is a gene that serves an important role in protein glycosylation and is essential for cancer cell metabolism. Administration of 2DG decreases GFAT1 in a dose-dependent manner leading to disruption of protein N-glycosylation. This causes ER stress-induced cell death of cancer cells.
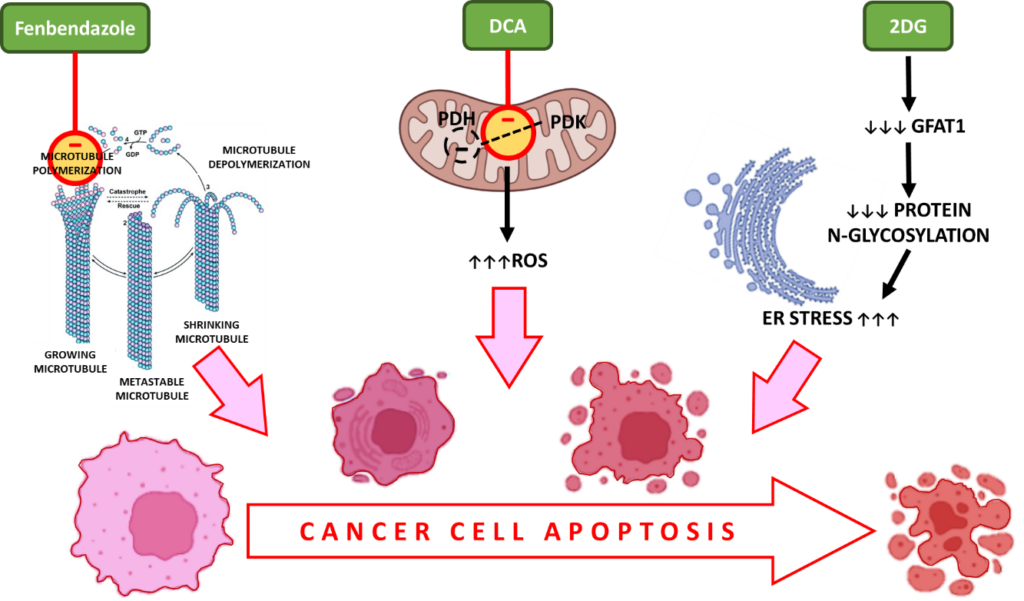
Inducing Apoptosis in Cancer with Fenbendazole, DCA and 2-DG
c) How the 3 drugs complement standard cancer treatments to exert further synergistic effects
i. Fenbendazole – low side-effect profile, therefore easy to co-administer with traditional treatments and obtain synergistic effects
Another important aspect of FBZ is its high safety margin, low toxicity and minimal drug resistance compared to other chemotherapeutic agents like taxanes and vinca alkaloids which also disrupts tubulin polymerization and arrests cell division but causes acute toxicity in humans.
In addition, FBZ also demonstrates reduction in new blood vessel formation via angiogenesis and inhibition of tumor growth in lung cancer cells.
ii. DCA – Reverses resistance to chemotherapy and radiotherapy
DCA treatment inhibits pyruvate dehydrogenase kinase (PDK). Decrease in PDH phosphorylation by PDK decreases phosphorylated pyruvate dehydrogenase (pPDH), lowers aerobic glycolysis, lactate production and extracellular acidification rate (ECAR).
By disrupting aerobic glycolysis DCA renders the cancer cells more sensitive to chemotherapeutic drugs while increased ROS imparts more radiation sensitivity to the cancer cells. A study performed in lung cancer and liver cancer cells showed that co administration of DCA with paclitaxel, a chemotherapeutic agent, increased apoptotic cell death in cancer cells.
iii. 2-DG – slows down metabolically overactive cancer cells and makes them more vulnerable to traditional treatments
2DG is also known to induce the expression of thioredoxin interacting protein (TXNIP), which is a tumor suppressor protein. By doing so it increases chemosensitivity and radiation sensitivity of the cancer cells.
This in turn results in increased apoptotic cell death in response to these conventional treatments. As such, 2-DG acts as a chemotherapy and radiation therapy sensitizer in cancer cells.
Combating Cancer with Fenbendazole, DCA, and 2-DG to Overcome Metabolic Reprogramming
The currently prevailing paradigm for cancer treatment largely depends on surgery, radiation therapy and chemotherapy. This traditionally employed combination approach has been successful in thwarting the progression of cancer in many cases and has even been successful in some cases of effectively curing patients.
However, for those patients who are either diagnosed at advanced and/or metastatic stages of disease, or those who unfortunately progress to advanced and/or metastatic stages of disease, the above combination approach has largely been ineffective. The reason is that cancer cells, when faced with imminent death due to therapeutic pressure, quite often can adapt through a process called Metabolic Reprogramming in which they learn to differentially utilize typical sources of cellular energy, like glucose, in a manner that makes them resistant to chemotherapy.
And until this metabolic adaptation is targeted, treatments will largely remain ineffective for advanced and/or metastatic cancers. Luckily safe and effective drugs to overcome Metabolic Reprogramming already exist and are currently available to cancer patients. Three such drugs include Fenbendazole, DCA and 2-DG which individually work through a variety of mechanisms.
When used in combination they often exert synergistic effects and can be even more effective than when used in a stand-alone manner. Furthermore, their use does not preclude utilization of standard treatments like chemotherapy or radiation therapy.
Based on the data available, administration of Fenbendazole, DCA and/or 2-DG in conjunction with traditional treatments is predicted to improve the overall effectiveness of cancer treatment, especially in the advanced and/or metastatic setting.
Future Outlook
While clinical studies and trials are ongoing to better understand how these 3 drugs can be further utilized for therapeutic benefit, they are currently available for patients today to be combined in treatment regimens.
References:
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021 May;71(3):209-249. doi: 10.3322/caac.21660. Epub 2021 Feb 4. PMID: 33538338.
- Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022 Jan;12(1):31-46. doi: 10.1158/2159-8290.CD-21-1059. PMID: 35022204.
- Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019 Nov;575(7782):299-309. doi: 10.1038/s41586-019-1730-1. Epub 2019 Nov 13. PMID: 31723286; PMCID: PMC8008476.
- Bukowski K, Kciuk M, Kontek R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int J Mol Sci. 2020 May 2;21(9):3233. doi: 10.3390/ijms21093233. PMID: 32370233; PMCID: PMC7247559.
- Nong S, Han X, Xiang Y, Qian Y, Wei Y, Zhang T, Tian K, Shen K, Yang J, Ma X. Metabolic reprogramming in cancer: Mechanisms and therapeutics. MedComm (2020). 2023 Mar 27;4(2):e218. doi: 10.1002/mco2.218. PMID: 36994237; PMCID: PMC10041388.
- Vaupel P, Multhoff G. Revisiting the Warburg effect: historical dogma versus current understanding. J Physiol. 2021 Mar;599(6):1745-1757. doi: 10.1113/JP278810. Epub 2021 Jan 4. PMID: 33347611.
- Batch JT, Lamsal SP, Adkins M, Sultan S, Ramirez MN. Advantages and Disadvantages of the Ketogenic Diet: A Review Article. Cureus. 2020 Aug 10;12(8):e9639. doi: 10.7759/cureus.9639. PMID: 32923239; PMCID: PMC7480775.
- Fu L, Jin W, Zhang J, Zhu L, Lu J, Zhen Y, Zhang L, Ouyang L, Liu B, Yu H. Repurposing non-oncology small-molecule drugs to improve cancer therapy: Current situation and future directions. Acta Pharm Sin B. 2022 Feb;12(2):532-557. doi: 10.1016/j.apsb.2021.09.006. Epub 2021 Sep 10. PMID: 35256933; PMCID: PMC8897051.
- Dogra N, Kumar A, Mukhopadhyay T. Fenbendazole acts as a moderate microtubule destabilizing agent and causes cancer cell death by modulating multiple cellular pathways. Sci Rep. 2018 Aug 9;8(1):11926. doi: 10.1038/s41598-018-30158-6. PMID: 30093705; PMCID: PMC6085345.
- Park D, Lee JH, Yoon SP. Anti-cancer effects of fenbendazole on 5-fluorouracil-resistant colorectal cancer cells. Korean J Physiol Pharmacol. 2022 Sep 1;26(5):377-387. doi: 10.4196/kjpp.2022.26.5.377. PMID: 36039738; PMCID: PMC9437363.
- Song B, Park EY, Kim KJ, Ki SH. Repurposing of Benzimidazole Anthelmintic Drugs as Cancer Therapeutics. Cancers (Basel). 2022 Sep 22;14(19):4601. doi: 10.3390/cancers14194601. PMID: 36230527; PMCID: PMC9559625.
- Tataranni T, Piccoli C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxid Med Cell Longev. 2019 Nov 14;2019:8201079. doi: 10.1155/2019/8201079. PMID: 31827705; PMCID: PMC6885244.
- Parczyk J, Ruhnau J, Pelz C, Schilling M, Wu H, Piaskowski NN, Eickholt B, Kühn H, Danker K, Klein A. Dichloroacetate and PX-478 exhibit strong synergistic effects in a various number of cancer cell lines. BMC Cancer. 2021 Apr 30;21(1):481. doi: 10.1186/s12885-021-08186-9. PMID: 33931028; PMCID: PMC8086110.
- O’Neill S, Porter RK, McNamee N, Martinez VG, O’Driscoll L. 2-Deoxy-D-Glucose inhibits aggressive triple-negative breast cancer cells by targeting glycolysis and the cancer stem cell phenotype. Sci Rep. 2019 Mar 7;9(1):3788. doi: 10.1038/s41598-019-39789-9. PMID: 30846710; PMCID: PMC6405919.
- Pajak B, Siwiak E, Sołtyka M, Priebe A, Zieliński R, Fokt I, Ziemniak M, Jaśkiewicz A, Borowski R, Domoradzki T, Priebe W. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int J Mol Sci. 2019 Dec 29;21(1):234. doi: 10.3390/ijms21010234. PMID: 31905745; PMCID: PMC6982256.
- Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014 Dec 28;355(2):176-83. doi: 10.1016/j.canlet.2014.09.003. Epub 2014 Sep 10. PMID: 25218591.

{kind=link}